
Cell and Gene therapies: Technology and opportunities
Brian Mullan (Yposkesi).
Keywords
Cell therapies, gene therapies, manufacturing, challenges, CMC.
Abstract
Cell and gene therapies (C>) are a fast growing and promising class of medicines that offer the possibility of curing conditions with a genetic basis that have few or no other therapeutic options for patients. From a turbulent start in the 1990s, with some notable setbacks in clinical trials, the promise of these therapies has now become a reality, with numerous market approvals in recent years, a robust clinical pipeline and strong industry and investor interest. The sector is both immature, from the regulatory and CMC perspectives, and growing fast. Numerous challenges will need to be surmounted in the coming years to ensure that these therapies can reliably reach patients, but the future looks very promising.
Introduction: What are cell and gene therapies?
Cell and Gene Therapies (C>) are a growing category of medicines that enable genetic defects to be directly corrected in patients, thus enabling remediation of disease symptoms that typically have no other therapeutic option (1). Some of the diseases treated by C> have existing small or large molecule therapeutic options (e.g., Spinraza (2) from Biogen/Ionis for Spinal Muscular Atrophy, SMA, which can also be treated by the C> Zolgensema (3) from Novartis), but for many of the target conditions C> often represents the only treatment option for patients.
C> work by providing a functioning/non-defective copy of a gene directly to the patient, which can enable the relevant underlying biology to function correctly, thus remedying the symptoms caused by the genetic defects (1). As such, these therapies provide a cure, as opposed to an ongoing treatment. There is also a growing sub-category of C> products based on gene-editing approaches (4), whereby the C> can repair or edit the defective gene, as opposed to substituting it.
The therapeutic approach for C> typically targets “monogenic” diseases, where the clinical symptoms are due to a defect in one specific gene. Monogenic disorders include metabolic conditions, e.g., defective enzymes, and physiological conditions such as Duchenne’s muscular dystrophy, which has serious effects on normal muscular development. Another sub-category of C> works via immune stimulation approaches where a novel protein is expressed to trigger an anti-disease response by the patient’s immune system (e.g., Chimeric Antigenic Receptor T-cell, or CAR-T, therapeutics (5)).
The replacement gene (or novel protein) can be delivered in two principal ways to the patient (1):
• in vivo: the gene of interest or the genetic editing machinery is packaged in to a viral vector (such as an Adeno Associated Virus, or AAV; 6) and injected directly into the patient as a parenteral medicine. A formulated solution of the viral vector constitutes the drug product. Non-viral approaches (such as plasmid DNAs encoding the gene of interest) are also possible, but are less prevalent (fewer products are in clinical development). The specific sub-type (or “serotype”) of the viral vector, and the genetic elements included in the vector, confer a degree of specificity to the vector, enabling it to target certain organs or tissues. The administration route can also enable this (e.g., ocular as opposed to systemic injections);
• ex vivo: a blood sample is taken from the patient, and a specific target cell of therapeutic interest is then isolated from the blood. The isolated cells are then “infected” (transduced) by a viral vector carrying the gene of interest or the genetic editing machinery, the cells amplified (to increase their number), further processed, and then re-injected back in to the patient (1). Lentiviral vectors (LVV) (7) are the most common vector used for this approach. The final product is the ex vivo transduced cell and is considered as a cell therapy (the viral vector used to transduce the target cells is considered as a critical starting material). These products are mainly used to treat blood cancers, where targeting specific genes or providing a specific immune stimulus (e.g., via a CAR-T; 5) provides a viable treatment option for patients.
Early years
Research and development of cell and gene therapies has been ongoing since the 1990s. For gene therapies (delivery of genes or gene-editing machinery using viral vectors), three principle types of vector have figured prominently: the aforementioned AAV and LVV, and Adenoviruses (AdV). The latter has somewhat fallen out of favour as a gene therapy vector, principally due to immune response issues (many patients have pre-existing immune responses to AdV), among other considerations, but are still widely used as vaccines (e.g., the current Astra Zeneca and Janssen COVID vaccines use Adenovirus as a vaccine vector) and as oncolytic viruses.
Many clinical trials were conducted in 1990s to progress viral vector-based gene therapies towards the market. Some notable examples include the X-linked severe combined immunodeficiency (SCID-X1 or “bubble boy”) treatment (8). However, some issues occurred during these early clinical trials, most prominently with the Gelsinger case in 1999 (9). The effect of this was to slow down the overall clinical development of the product class.
Market growth
Development nonetheless continued, focusing on AAV and LVVs as principal vectors, and in the 2010’s, the first full market approvals started to happen (10), with 19 therapies (10, 11) now marketed as of Q3 2021. In total, >2000 C> are in development as of Q3 2021 (10, 11) and ~1220 clinical trials are ongoing, with ~30 in Phase III (10, 11). These include ~400 in vivo gene therapies and ~800 cell therapies, the latter of which can be broadly split into two categories: autologous therapies, wherein the patients’ own cells are modified, and used to treat only that patient, and allogenic therapies, where donor cells are modified and used for multiple patients (1).
There has been over 20 Billion USD invested in the sector in 2020 (11). Numerous new companies have been created to progress these therapies to the clinic, as well as the entry of established pharma players in to the sector (12).
The sector is forecast to grow strongly in the coming years (12, 13).
Manufacturing process overview
A generic overview of typical C> manufacturing process is presented in Figure 1:
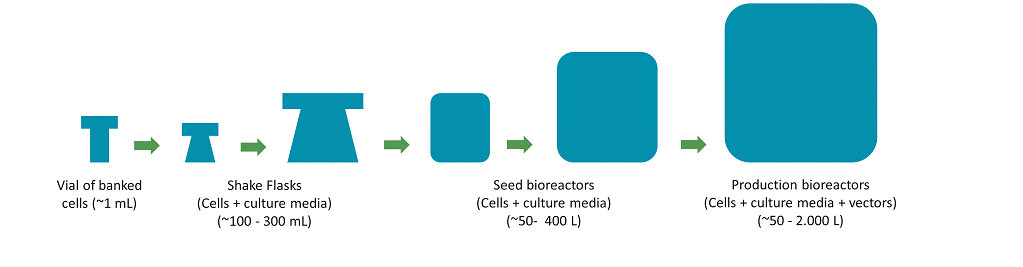
Figure 1a. Generic schema for a gene therapy viral vector mammalian cell manufacturing process (First part – cell culture): mammalian cells are progressively expanded in containers of increasing size, until enough culture mass and viability is obtained to start a Production Bioreactor.
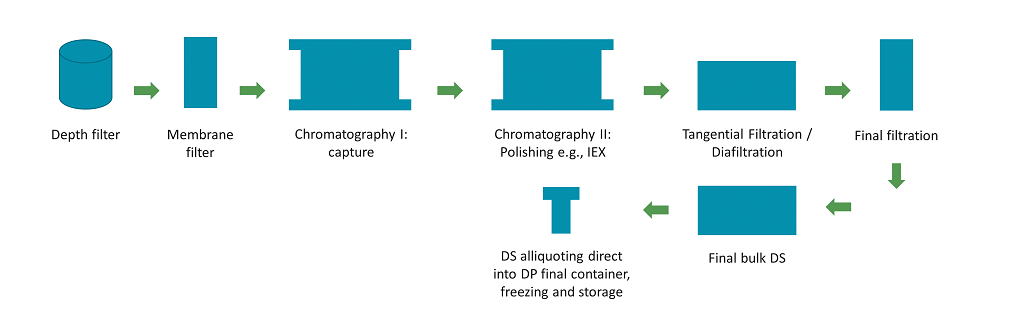
Figure 1b. Generic schema for a gene therapy viral vector mammalian cell manufacturing process (Second part – purification). Cells and cell debris are removed from the crude harvest via filtration. The clarified harvest is then processed through a series of chromatography and filtration steps to separate the product of interest from impurities. A 0.2um filtration step (not shown here) is usually included between each unit operation for bioburden control.
Gene therapy viral vector manufacturing processes are very different to those used for small molecules/chemistry-based therapeutics, but highly similar to manufacturing processes used for therapeutic monoclonal antibodies (MAb), recombinant proteins, and vaccines. In essence, mammalian cell lines are grown up to a sufficient quantity/volume, and used to produce the viral vector. The vector can be introduced in to the producer cells by transient transfection of plasmid DNAs, or a packaging cell line can be used. A crude harvest (cells and vector) is obtained, which is then purified through different separation modalities to result in a final purified product (vector in a formulated buffer) (Figure 1).
Cell therapy (gene modified) manufacturing processes are more novel, and essentially involve selection and separation of target cells (from the patient), putting them in to contact with a viral vector (which enables the cells to be genetically modified), and then further purifying the intermediate to remove unused vector and ensure the final product is in an appropriate matrix (1). These processes by nature are very manual, and highly variable for autologous products, as the starting material is unique to each patient. Speed is of the essence, and in-process and final sampling needs to be minimized to avoid requirements for large blood samples from the patients, or excessive product losses due to sampling.
Current state
As regards the product and process development, or chemistry, manufacturing and control (CMC) aspects of these therapies, the overall C> industry is very immature but evolving rapidly. The state of understanding of CMC for viral vector development and manufacturing is similar to that for MAbs in the 1990s. For cell therapies, the manufacturing approaches are novel and there are few similarities to existing, well-established, pharmaceutical product classes). Some key CMC challenges for C> include:
• Product understanding: the overall understanding of mechanism of action, pharmacology, etc, is low. Issues still arise during clinical evaluation (14,15) which can lead to clinical holds that also affect similar products under development. As such, clinical development can be somewhat “stop-start”, while the industry learns from these events and tries continuously to ensure product safety and an appropriate risk-benefit balance. Linked to this is an evolving regulatory landscape. Recent communications from the FDA as regards the number of “empty vectors” in the final DP for AAVs is one example (16). In this specific case, the FDA highlighted a concern around potentially high dosing, and raised concerns about the proportion of “empty vectors” (not containing the genetic element) in AAV preparations. The agency requested that these are actively monitored, to encourage higher levels of “full” capsids in the final product, in order to reduce the required dose (16);
• Speed to market: as many C> often represent the only, or last line, therapeutic option for patients with time-critical conditions, clinical development timelines are typically accelerated, with clinical phases often combined. This can create challenges as regards the time required to gain appropriate product understanding to ensure right-first time regulatory submissions. Dedicated accelerated regulatory programmes have been established by Health Authorities, e.g., RMAT for the FDA (17) and PRIME for EMA (18), to support product developers. Additionally, other groups are working on platform approaches for certain vectors, where the principal difference between the products is the gene of interest (the viral vector is the same), and thus prior knowledge could be leveraged between development programmes (e.g., PAVE-GT by NIH (19), and BioPhorum (20));
• Low numbers of batches: due to the accelerated timelines, many manufacturing processes may only have a handful of manufactured batches in total prior to starting process performance qualification (PPQ). This makes CMC package development (product and process development and characterization, and product specification setting) very challenging. Accelerated programmes such as RMAT and PRIME (17, 18) enable ongoing dialogue between Health Authorities and product sponsors to navigate this. This may also mean that certain aspects of process or product characterisation and performance qualification may be extended in to the marketed phase of the product. Health Authorities appear to be requiring more and more that product understanding criteria are respected for each clinical development phase, thus ensuring a balance of safety vs patient access, as opposed to accepting that speed to patient / market is the predominant concern. A clear rationale of science versus risk is required;
• Understanding of product quality attributes: for C> this is quite rudimentary compared to other product classes. For the example of empty/full AAV capsids (21) mentioned previously (16), it is clear that empty AAV (not carrying the gene of interest) should not provide a therapeutic contribution, but the situation may not be unequivocal. Is there a possibility that empty capsids contribute to the overall pharmacology of the product? Similarly, guidance on targets / specifications for well-known process impurities, such as host cell proteins and DNA, are not yet established despite the well-known immunogenic nature of these impurities;
• Weak analytics: the nature of C> products are large and complex (AAV are ~25nm/3800kDa and LVV or ~100nm/200 MDa in size, respectively; e.g., compared to a MAb at ~150kDa), rendering specific and unambiguous analytical evaluation highly challenging. The situation with MAbs (as a comparator) already relies heavily on orthogonal analytics (multiple, complimentary analytical methods, that collectively provide an appropriate analytical evaluation of the product), as compared to the analytic power normally available for small molecule therapies;
• Manual processes, and the use of animal-derived materials: these aspects are very prevalent in cell therapy production processes, where many processes use largely manual processes to manipulate the patient cells and viral vectors. Due to the nature of the patient cells (ex vivo, often from sick patients), fully chemically defined raw / starting materials are not always possible to use (the cells need richer matrixes to survive the processing steps). They may require the use of raw materials such as calf / bovine sera and biological growth factors, which are generally not recommended for pharmaceutical manufacturing processes as they present potential safety risks and also create manufacturing variability.
Opportunities from chemical manufacturing
For gene therapy manufacturing processes, the production steps are highly similar to those used for MAbs, and the continued evolution of GT processes will most likely evolve in this sense. Specific areas of development for GT include chromatography resins and filtration, as the products are larger and more complex, and the separation chemistries and appropriate filtration matrices and approaches are not always identical to those used for MAbs. This may be an area where cross-pollination of technologies from chemical manufacturing would be beneficial. For (gene modified) cell therapies, most of the future progress will most likely come in two areas: 1) reduction where possible of the use of complex and/or animal derived raw materials, such as growth factors and culture media, and 2) continued automation of process handling steps. There appears to be less scope for cross-pollination with chemical manufacturing technologies here. The future for this class of therapies is extremely promising and inspiration and innovation will undoubtedly come from many different areas to continue the technological evolution.
References
1. High K.A., Roncarolo M.G. Gene Therapy. N. Engl. J. Med., 381(5), 455-464 (2019).
2. https://www.spinraza.com/ (last checked on Jan. 7th 2022).
3. https://www.zolgensma.com/ (last checked on Jan. 7th 2022).
4. Gene-editing pipeline takes off. Nat. Rev. Drug Discov., 19, 367-372 (2020).
5. Sadelain M., et al. The basic principles of chimeric antigen receptor design. Cancer Discov., 3, 388–398 (2013).
6. Wang D et al. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov., 18, 358 – 378 (2019).
7. Lynch, M., Yposkesi signs with Servier to manufacture allogenic CAR-T, Biopharma-Reporter, 11 June 2019, https://www.biopharma-repor ter.com/Article/2019/06/11/Servier-taps-Ypokeski-to-produce-lentiviral-vectors (last checked on Jan. 7th 2022)
8. Hacein-Bey-Abina S., et al. Sustained Correction of X-Linked Severe Combined Immunodeficiency by ex Vivo Gene Therapy. N. Engl. J. Med., 346,1185-1193 (2002).
9. Stolberg S. G. The biotech death of Jesse Gelsinger. New York Times Magazine. November 28th, 1999. http://www.nytimes.com/1999/11/28/magazine/the-biotech-death-of-jesse-gelsinger.html (last checked on Jan. 7th 2022)
10. American Society for Cell and Gene Therapy. Q3 2021 Quarterly Data Report.
11. Alliance for Regenerative Medicine, Annual Report 2020: Growth & Resilience in Regenerative medicine.
12. Gene therapy boom continues. Nat. Rev. Drug Discov., 19, 737 (2019).
13. Roots Analysis, Business Research and Consulting, Gene Therapy Market (4th Edition) by Therapeutic Approach (Gene Augmentation, Oncolytic Viral Therapy, Immunotherapy and Others), Type of Gene Therapy, Type of Vectors Used, Therapeutic Areas (Autoimmune Disorders, Cardiovascular Diseases, Genetic Disorders, Haematological Disorders, Metabolic Disorders, Muscle-related Diseases, Oncological Disorders, Ophthalmic Diseases and Others), Route of Administration, and Key Geographical Regions: Industry Trends and Global Forecasts, 2020-2030
14. Lentiviral vector cleared of causing blood cancer. Nat. Biotechnol., 39, 398 (2021).
15. Gene therapy community grapples with toxicity issues, as pipeline matures. Nat. Rev. Drug Discov., 20, 804-805 (2021).
16. Gene Therapy Dosing Caps Not Recommended By FDA Advisory Committee, But Standards Urged To Address “Empty” Capsids And Potential Toxicity. Prevision Policy. September 7th, 2021. https://www.previsionpolicy.com/gene-therapy-dosing-caps-not-recommended-by-fda-advisory-committee-but-standards-urged-to-address-empty-capsids-and-potential-toxicity (last checked on Jan. 7th 2022)
17. FDA. Regenerative Medicine Advanced Therapy Designation. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/regenerative-medicine-advanced-therapy-designation (last checked on Jan. 7th 2022)
18. EMA. PRIME: priority medicines. https://www.ema.europa.eu/en/human-regulatory/research-development/prime-priority-medicines (last checked on Jan. 7th 2022)
19. Brooks P. J., et al. The Platform Vector Gene Therapies Project: Increasing the Efficiency of Adeno Associated Virus Gene Therapy Clinical Trial Startup. Human Gene Therapy, 31(19-20), 1034 – 1042 (2020).
20. https://www.biophorum.com/ (last checked on Jan. 7th 2022)
21. Mietzsch M. et al. Improved Genome Packaging Efficiency of Adeno associated Virus Vectors Using Rep Hybrids. J. Virology. 95(19), doi:
10.1128/JVI.00773-21.
Copyright: Published by tks, publisher, event organiser media agency. January/February 2022 VOL. 40(1)